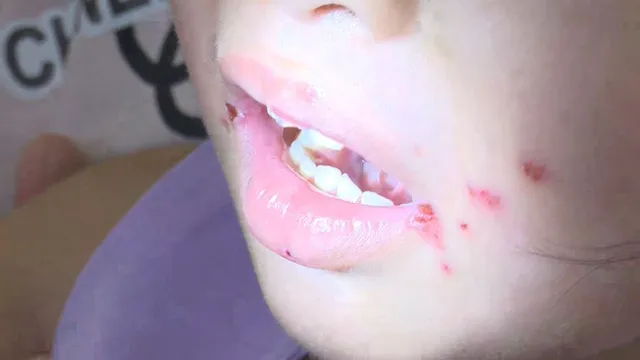
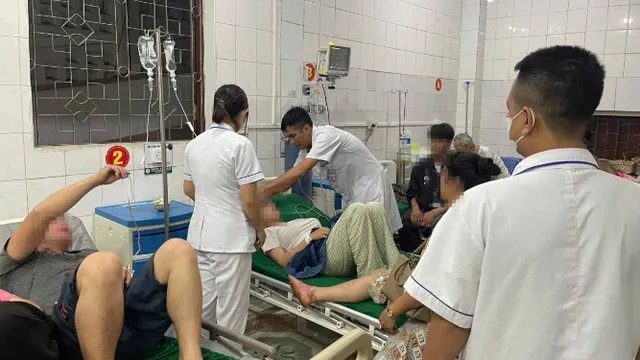
Các bác sĩ Khoa Huyết học lâm sàng và Bệnh nghề nghiệp - Bệnh viện Quân y 175 đã điều trị thành công một trường hợp Hội chứng Evans - bệnh rối loạn máu tự miễn cực hiếm - cho nữ bệnh nhân 17 tuổi, cư trú tại huyện Củ Chi, TP Hồ Chí Minh. Bệnh nhân nhập viện trong tình trạng thiếu máu nặng, xuất huyết dưới da, đau tức ngực và khó thở.
Theo hồ sơ bệnh án, bệnh nhân có tiền sử mẹ mắc giảm tiểu cầu miễn dịch. Trong 3 năm gần đây, bệnh nhân thường xuyên hoa mắt, chóng mặt, rong kinh kéo dài và được chẩn đoán thiếu máu thiếu sắt. Tuy nhiên, tình trạng không cải thiện sau điều trị. Một tuần trước khi nhập viện, các nốt xuất huyết bắt đầu lan khắp cơ thể, kèm đau ngực, khó thở, buộc phải cấp cứu.
Tại thời điểm tiếp nhận, bệnh nhân có huyết sắc tố chỉ 31g/L, tiểu cầu 8G/L – mức nguy hiểm tính mạng. Kết quả xét nghiệm chuyên sâu xác định đây là trường hợp Hội chứng Evans nguyên phát – một dạng rối loạn tự miễn khiến cơ thể tự sinh kháng thể tấn công hồng cầu, tiểu cầu, thậm chí cả bạch cầu.
Phác đồ điều trị được kích hoạt khẩn trương: truyền máu, sử dụng corticoid và thuốc ức chế miễn dịch. Sau 11 ngày điều trị tích cực, các chỉ số huyết học cải thiện rõ rệt, huyết sắc tố tăng lên 115g/L, tiểu cầu đạt 177G/L. Bệnh nhân được xuất viện trong tình trạng ổn định, tiếp tục theo dõi ngoại trú.
Theo bác sĩ Mai Thị Thu Ly, người trực tiếp điều trị ca bệnh, Hội chứng Evans chỉ chiếm khoảng 7% các ca tan máu tự miễn và 2% giảm tiểu cầu miễn dịch. Do triệu chứng ban đầu không điển hình, bệnh dễ bị nhầm với thiếu máu thông thường, rối loạn đông máu hoặc bệnh lý gan, khiến việc điều trị chậm trễ và có thể để lại biến chứng nguy hiểm.
"Hiện chưa có phác đồ chuẩn mực cho hội chứng này. Việc điều trị chủ yếu dựa trên kinh nghiệm và sự phối hợp linh hoạt giữa các chuyên khoa. Thành công điều trị bệnh nhân 17 tuổi vừa qua cho thấy năng lực xử trí hiệu quả của Bệnh viện Quân y 175 với các bệnh hiếm và phức tạp," bác sĩ Thu Ly nhấn mạnh.
Hội chứng Evans là rối loạn tự miễn hiếm gặp, đặc trưng bởi sự kết hợp giữa tan máu tự miễn (AIHA) và giảm tiểu cầu miễn dịch (ITP), đôi khi kèm giảm bạch cầu. Bệnh có thể đơn phát hoặc liên quan đến các bệnh tự miễn khác như lupus ban đỏ hệ thống, hội chứng Sjogren, hoặc rối loạn lympho.
Triệu chứng bao gồm da xanh, mệt mỏi, sốt, xuất huyết dưới da, vàng da, chảy máu chân răng. Gan, lách có thể to. Chẩn đoán dựa trên lâm sàng và các chỉ số xét nghiệm như Coombs dương tính, giảm hemoglobin và tiểu cầu, tăng bilirubin gián tiếp.
Điều trị khởi đầu bằng corticosteroid, globulin miễn dịch. Các trường hợp kháng trị có thể dùng rituximab, cyclosporin A hoặc cắt lách. Do bệnh lý phức tạp, việc theo dõi sát và cá thể hóa điều trị là yếu tố quyết định hiệu quả lâu dài.